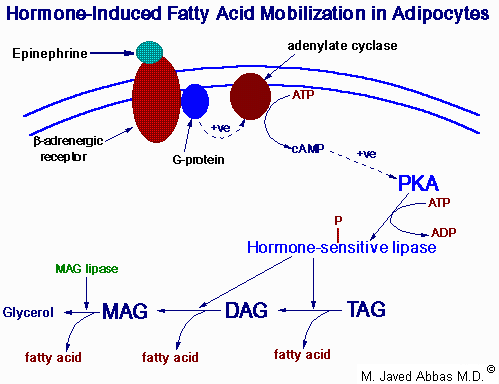
| Model for the activation of hormone-sensitive lipase by epinephrine. Epinephrine binds its' receptor and leads to the activation of adenylate cyclase. The resultant increase in cAMP activates PKA which then phosphorylates and activates hormone-sensitive lipase. Hormone-sensitive lipase hydrolyzes fatty acids from triacylglycerols and diacylglycerols. The final fatty acid is released from monoacylglycerols through the action of monoacylglycerol lipase, an enzyme active in the absence of hormonal stimulation.
|