Introduction
The ability of the body to control the flow of blood following vascular injury is paramount to continued survival. The process of blood clotting and then the subsequent dissolution of the clot, following repair of the injured tissue, is termed hemostasis. Hemostasis, composed of 4 major events that occur in a set order following the loss of vascular integrity:
- 1. The initial phase of the process is vascular constriction. This limits the flow of blood to the area of injury.
- 2. Next, platelets become activated by thrombin and aggregate at the site of injury, forming a temporary, loose platelet plug. The protein fibrinogen is primarily responsible for stimulating platelet clumping. Platelets clump by binding to collagen that becomes exposed following rupture of the endothelial lining of vessels. Upon activation, platelets release adenosine-5'-diphosphate, ADP and TXA2 (which activate additional platelets), serotonin, phospholipids, lipoproteins, and other proteins important for the coagulation cascade. In addition to induced secretion, activated platelets change their shape to accommodate the formation of the plug.
- 3. To insure stability of the initially loose platelet plug, a fibrin mesh (also called the clot) forms and entraps the plug. If the plug contains only platelets it is termed a white thrombus; if red blood cells are present it is called a red thrombus.
- 4. Finally, the clot must be dissolved in order for normal blood flow to resume following tissue repair. The dissolution of the clot occurs through the action of plasmin.
Two pathways lead to the formation of a fibrin clot: the intrinsic and extrinsic pathway. Although they are initiated by distinct mechanisms, the two converge on a common pathway that leads to clot formation. The formation of a red thrombus or a clot in response to an abnormal vessel wall in the absence of tissue injury is the result of the intrinsic pathway. Fibrin clot formation in response to tissue injury is the result of the extrinsic pathway. Both pathways are complex and involve numerous different proteins termed clotting factors.
back to the top
Platelet Activation and von Willebrand Factor (vWF)
In order for hemostasis to occur, platelets must adhere to exposed collagen, release the contents of their granules, and aggregate. The adhesion of platelets to the collagen exposed on endothelial cell surfaces is mediated by von Willebrand factor (vWF). Inherited deficiencies of vWF are the causes of von Willebrand disease, (vWD) (see below for more details). The function of vWF is to act as a bridge between a specific glycoprotein on the surface of platelets (GPIb/IX) and collagen fibrils. In addition to its role as a bridge between platelets and exposed collagen on endothelial surfaces, vWF binds to and stabilizes coagulation factor VIII. Binding of factor VIII by vWF is required for normal survival of factor VIII in the circulation.
von Willebrand factor is a complex multimeric glycoprotein that is produced by and stored in the a-granules of platelets. It is also synthesized by megakaryocytes and found associated with subendothelial connective tissue.
The initial activation of platelets is induced by thrombin binding to specific receptors on the surface of platelets, thereby initiating a signal transduction cascade. The thrombin receptor is coupled to a G-protein that, in turn, activates phospholipase C-g (PLC-g). PLC-g hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2) leading to the formation of inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 induces the release of intracellular Ca2+ stores, and DAG activates protein kinase C (PKC).
The collagen to which platelets adhere as well as the release of intracellular Ca2+ leads to the activation of phospholipase A2 (PLA2), which then hydrolyzes membrane phospholipids, leading to liberation of arachidonic acid. The arachidonic acid release leads to an increase in the production and subsequent release of thromboxane A2 (TXA2). This is another platelet activator that functions through the PLC-g pathway. Another enzyme activated by the released intracellular Ca2+ stores is myosin light chain kinase (MLCK). Activated MLCK phosphorylates the light chain of myosin which then interacts with actin, resulting in altered platelet morphology and motility.
One of the many effects of PKC is the phosphorylation and activation of a specific 47,000-Dalton platelet protein. This activated protein induces the release of platelet granule contents; one of which is ADP. ADP further stimulates platelets increasing the overall activation cascade; it also modifies the platelet membrane in such a way as to allow fibrinogen to adhere to two platelet surface glycoproteins, GPIIb and GPIIIa, resulting in fibrinogen-induced platelet aggregation.
Activation of platelets is required for their consequent aggregation to a platelet plug. However, equally significant is the role of activated platelet surface phospholipids in the activation of the coagulation cascade.
back to the top
Primary Factors
| Factor | Trivial Name(s) | Pathway | Characteristic |
| Prekallikrein | Fletcher factor | Intrinsic | |
| High molecular weight kininogen (HMWK) | contact activation cofactor; Fitzgerald, Flaujeac Williams factor | Intrinsic | |
| I | Fibrinogen | Both | - |
| II | Prothrombin | Both | Contains N-term. gla segment |
| III | Tissue Factor | Extrinsic | - |
| IV | Calcium | Both | - |
| V | Proaccelerin, labile factor, accelerator (Ac-) globulin | Both | Protein cofactor |
| VI (Va) | Accelerin | - | This is Va, redundant to Factor V |
| VII | Proconvertin, serum prothrombin conversion accelerator (SPCA), cothromboplastin | Extrinsic | Endopeptidase with gla residues |
| VIII | Antihemophiliac factor A, antihemophilic globulin (AHG) | Intrinsic | Protein cofactor |
| IX | Christmas Factor,
antihemophilic factor B,plasma thromboplastin component (PTC) | Intrinsic | Endopeptidase with gla residues |
| X | Stuart-Prower Factor | Both | Endopeptidase with gla residues |
| XI | Plasma thromboplastin antecedent (PTA) | Intrinsic | Endopeptidase |
| XII | Hageman Factor | Intrinsic | Endopeptidase |
| XIII | Protransglutaminase,
fibrin stabilizing factor (FSF), fibrinoligase | Both | Transpeptidase |
Functional Classification of Clotting Factors
| Zymogens of Serine Proteases | Activities |
| Factor XII | binds to exposed collagen at site of vessel wall injury, activated by high-MW kininogen and kallikrein |
| Factor XI | activated by factor XIIa |
| Factor IX | activated by factor XIa in presence of Ca2+ |
| Factor VII | activated by thrombin in presence of Ca2+ |
| Factor X | activated on surface of activated platelets by tenase complex and by factor VIIa in presence of tissue factor and Ca2+ |
| Factor II | activated on surface of activated platelets by prothrombinase complex |
| Cofactors | Activities |
| Factor VIII | activated by thrombin; factor VIIIa is a cofactor in the activation of factor X by factor IXa |
| Factor V | activated by thrombin; factor Va is a cofactor in the activation of prothrombin by factor Xa |
| Factor III (tissue factor) | a subendothelial cell-surface glycoprotein that acts as a cofactor for factor VII |
| Fibrinogen | Activity |
| Factor I | cleaved by thrombin to form fibrin clot |
| Transglutaminase | Activity |
| Factor XIII | activated by thrombin in presence of Ca2+; stabilizes fibrin clot by covalent cross-linking |
| Regulatory and other proteins | Activities |
| von Willebrand factor | associated with subendothelial connective tissue; serves as a brigde between platelet glycoprotein GPIb/IX and collagen |
| Protein C | activated to protein Ca by thrombin bound to thrombomodulin; then degrades factors VIIIa and Va |
| Protein S | acts as a cofactor of protein C; both proteins contain gla residues |
| Thrombomodulin | protein on the surface of endothelial cells; binds thrombin, which then activates protein C |
| Antithrombin III | most important coagulation inhibitor, controls activities of thrombin, and factors IXa, Xa, XIa and XIIa |
back to the top
The Clotting Cascades
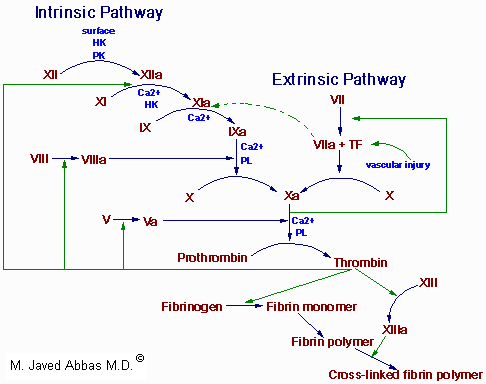 |
| The clotting cascades. The intrinsic cascade is initiated when contact is made between blood and exposed endothelial cell surfaces. The extrinsic pathway is initiated upon vascular injury which leads to exposure of tissue factor (TF) (also identified as factor III), a subendothelial cell-surface glycoprotein that binds phospholipid. The green dotted arrow represents a point of cross-over between the extrinsic and intrinsic pathways. The two pathways converge at the activation of factor X to Xa. Factor Xa has a role in the further activation of factor VII to VIIa as depicted by the green arrow. Active factor Xa hydrolyzes and activates prothrombin to thrombin. Thrombin can then activate factors XI, VIII and V furthering the cascade. Ultimately the role of thrombin is to convert fribrinogen to fibrin and to activate factor XIII to XIIIa. Factor XIIIa (also termed transglutaminase) cross-links fibrin polymers solidifying the clot. HK = high molecular weight kininogen. PK = prekallikrein. |
back to the top
The Intrinsic Clotting Cascade
The intrinsic pathway requires the clotting factors VIII, IX, X, XI, and XII. Also required are the proteins prekallikrein and high-molecular-weight kininogen, as well as calcium ions and phospholipids secreted from platelets. Each of these pathway constituents leads to the conversion of factor X (inactive) to factor Xa (a signifies active). Initiation of the intrinsic pathway occurs when prekallikrein, high-molecular-weight kininogen, factor XI and factor XII are exposed to a negatively charged surface. This is termed the contact phase. Exposure of collagen to a vessel surface is the primary stimulus for the contact phase.
The assemblage of contact phase components results in conversion of prekallikrein to kallikrein, which in turn activates factor XII to factor XIIa. Factor XIIa can then hydrolyze more prekallikrein to kallikrein, establishing a reciprocal activation cascade. Factor XIIa also activates factor XI to factor XIa and leads to the release of bradykinin, a potent vasodilator, from high-molecular-weight kininogen.
In the presence of Ca2+, factor XIa activates factor IX to factor IXa. Factor IX is a proenzyme that contains vitamin K-dependent g-carboxyglutamate (gla) residues, whose serine protease activity is activated following Ca2+ binding to these gla residues. Several of the serine proteases of the cascade (II, VII, IX, and X) are gla-containing proenzymes. Active factor IXa cleaves factor X at an internal arg-ile bond leading to its activation to factor Xa.
The activation of factor Xa requires assemblage of the tenase complex (Ca2+ and factors VIIIa, IXa and X) on the surface of activated platelets. One of the responses of platelets to activation is the presentation of phosphatidylserine and phosphatidylinositol on their surfaces. The exposure of these phospholipids allows the tenase complex to form. The role of factor VIII in this process is to act as a receptor, in the form of factor VIIIa, for factors IXa and X. Factor VIIIa is termed a cofactor in the clotting cascade. The activation of factor VIII to factor VIIIa (the actual receptor) occurs in the presence of minute quantities of thrombin. As the concentration of thrombin increases, factor VIIIa is ultimately cleaved by thrombin and inactivated. This dual action of thrombin, upon factor VIII, acts to limit the extent of tenase complex formation and thus the extent of the coagulation cascade.
back to the top
Extrinsic Clotting Cascade
Activated factor Xa is the site at which the intrinsic and extrinsic coagulation cascades converge. The extrinsic pathway is initiated at the site of injury in response to the release of tissue factor (factor III). Tissue factor is a cofactor in the factor VIIa-catalyzed activation of factor X. Factor VIIa, a gla residue containing serine protease, cleaves factor X to factor Xa in a manner identical to that of factor IXa of the intrinsic pathway. The activation of factor VII occurs through the action of thrombin or factor Xa.
The ability of factor Xa to activate factor VII creates a link between the intrinsic and extrinsic pathways. An additional link between the two pathways exists through the ability of tissue factor and factor VIIa to activate factor IX. The formation of complex between factor VIIa and tissue factor is believed to be a principal step in the overall clotting cascade. Evidence for this stems from the fact that persons with hereditary deficiencies in the components of the contact phase of the intrinsic pathway do not exhibit clotting problems.
A major mechanism for the inhibition of the extrinsic pathway occurs at the tissue factor--factor VIIa--Ca2+--Xa complex. The protein, lipoprotein-associated coagulation inhibitor, LACI specifically binds to this complex. LACI is also referred to as extrinsic pathway inhibitor, EPI or tissue factor pathway inhibitor, TFPI and was formerly named anticonvertin. LACI is composed of 3 tandem protease inhibitor domains. Domain 1 binds to factor Xa and domain 2 binds to factor VIIa only in the presence of factor Xa.
back to the top
Activation of Prothrombin to Thrombin
The common point in both pathways is the activation of factor X to factor Xa. Factor Xa activates prothrombin (factor II) to thrombin (factor IIa). Thrombin, in turn, converts fibrinogen to fibrin. The activation of thrombin occurs on the surface of activated platelets and requires formation of a prothrombinase complex. This complex is composed of the platelet phospholipids, phosphatidylinositol and phosphatidylserine, Ca2+, factors Va and Xa, and prothrombin. Factor V is a cofactor in the formation of the prothrombinase complex, similar to the role of factor VIII in tenase complex formation. Like factor VIII activation, factor V is activated to factor Va by means of minute amounts and is inactivated by increased levels of thrombin. Factor Va binds to specific receptors on the surfaces of activated platelets and forms a complex with prothrombin and factor Xa.
Prothrombin is a 72,000-Dalton, single-chain protein containing ten gla residues in its N-terminal region. Within the prothrombinase complex, prothrombin is cleaved at 2 sites by factor Xa. This cleavage generates a 2-chain active thrombin molecule containing an A and a B chain which are held together by a single disulfide bond.
In addition to its role in activation of fibrin clot formation, thrombin plays an important regulatory role in coagulation. Thrombin combines with thrombomodulin present on endothelial cell surfaces forming a complex that converts protein C to protein Ca. The cofactor protein S and protein Ca degrade factors Va and VIIIa, thereby limiting the activity of these 2 factors in the coagulation cascade.
Thrombin also binds to and leads to the release of G-protein-coupled protease activated receptors (PARs), specifically PAR-1, -3 and -4. The release of these proteins leads to the activation of numerous signaling cascades that in turn increase release of the interleukins, ILs, IL-1 and IL-6, increases secretion of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). The thrombin-induced signaling also leads to increased platelet activation and leukocyte adhesion. Thrombin also activates thrombin-activatable fibrinolysis inhibitor (TAFI) thus modulating fibrinolysis (degradation of fibrin clots). TAFI is also known as carboxypeptidase U (CPU) whose activity leads to removal of C-terminal lysines from partially degraded fibrin. This leads to an impairment of plasminogen activation, thereby reducing the rate of fibrin clot dissolution (i.e. fibrinolysis).
back to the top
Control of Thrombin Levels
The inability of the body to control the circulating level of active thrombin would lead to dire consequences. There are 2 principal mechanisms by which thrombin activity is regulated. The predominant form of thrombin in the circulation is the inactive prothrombin, whose activation requires the pathways of proenzyme activation described above for the coagulation cascade. At each step in the cascade, feedback mechanisms regulate the balance between active and inactive enzymes.
The activation of thrombin is also regulated by 4 specific thrombin inhibitors. Antithrombin III is the most important since it can also inhibit the activities of factors IXa, Xa, XIa and XIIa. The activity of antithrombin III is potentiated in the presence of heparin by the following means: heparin binds to a specific site on antithrombin III, producing an altered conformation of the protein, and the new conformation has a higher affinity for thrombin as well as its other substrates. This effect of heparin is the basis for its clinical use as an anticoagulant. The naturally occurring heparin activator of antithrombin III is present as heparan and heparan sulfate on the surface of vessel endothelial cells. It is this feature that controls the activation of the intrinsic coagulation cascade.
However, thrombin activity is also inhibited by a2-macroglobulin, heparin cofactor II and a1-antitrypsin. Although a minor player in thrombin regulation a1-antitrypsin is the primary serine protease inhibitor of human plasma. Its physiological significance is demonstrated by the fact that lack of this protein plays a causative role in the development of emphysema.
back to the top
Activation of Fibrinogen to Fibrin
Fibrinogen (factor I) consists of 3 pairs of polypeptides ([A-a][B-b][g])2. The 6 chains are covalently linked near their N-terminals through disulfide bonds. The A and B portions of the A-a and B-b chains comprise the fibrinopeptides, A and B, respectively. The fibrinopeptide regions of fibrinogen contain several glutamate and aspatate residues imparting a high negative charge to this region and aid in the solubility of fibrinogen in plasma. Active thrombin is a serine protease that hydrolyses fibrinogen at four arg-gly bonds between the fibrinopeptide and the a and b portions of the protein.
Thrombin-mediated release of the fibrinopeptides generates fibrin monomers with a subunit structure (a-b- g)2. These monomers spontaneously aggregate in a regular array, forming a somewhat weak fibrin clot. In addition to fibrin activation, thrombin converts factor XIII to factor XIIIa, a highly specific transglutaminase that introduces cross-links composed of covalent bonds between the amide nitrogen of glutamines and e-amino group of lysines in the fibrin monomers.
back to the top
Dissolution of Fibrin Clots
Degradation of fibrin clots is the function of plasmin, a serine protease that circulates as the inactive proenzyme, plasminogen. Any free circulating plasmin is rapidly inhibited by a2-antiplasmin. Plasminogen binds to both fibrinogen and fibrin, thereby being incorporated into a clot as it is formed. Tissue plasminogen activator (tPA) and, to a lesser degree, urokinase are serine proteases which convert plasminogen to plasmin. Inactive tPA is released from vascular endothelial cells following injury; it binds to fibrin and is consequently activated. Urokinase is produced as the precursor, prourokinase by epithelial cells lining excretory ducts. The role of urokinase is to activate the dissolution of fibrin clots that may be deposited in these ducts.
Active tPA cleaves plasminogen to plasmin which then digests the fibrin; the result is soluble degradation product to which neither plasmin nor plasminogen can bind. Following the release of plasminogen and plasmin they are rapidly inactivated by their respective inhibitors. The inhibition of tPA activity results from binding to specific inhibitory proteins. At least 4 distinct inhibitors have been identified, of which 2-- plasminogen activator-inhibitors type 1 (PAI-1) and type 2 (PAI-2) are of greatest physiological significance.
back to the top
Clinical Significances of Hemostasis:
The Bleeding Disorders
Defects in the process of hemostasis, leading to bleeding disorders, have been identified at the level of the proteins of the clotting cascades, platelet activation and function, contact activation and antithrombin function.
Hemophilia A
Hemophilia A is classic hemophilia (a disease referring to the inability to clot blood). It is an X-linked disorder resulting from a deficiency in factor VIII, a key component of the coagulation cascade. There are severe, moderate and mild forms of hemophilia A that reflect the level of active factor VIII in the plasma.
Hemophilia A arises from a variety of mutations. Some 150 different point mutations have been characterized in the factor VIII gene in hemophilia A. Inheritence of the disorder occurs with a frequency of 1:5,000 to 1:10,000 males in all populations. Factor VIII is a cofactor in the activation of factor X to factor Xa in a reaction catalyzed by factor IXa. Activation of factor VIII occurs via proteolytic cleavage by thrombin and factor Xa. Inactivaqtion of factor VIIIa occurs by limited proteolysis by factor Xa or activated protein C.
Individuals with deficiencies in factor VIII suffer joint and muscle hemorrhage, easy bruising and prolonged bleeding from wounds. Treatment of hemophilia A is accomplished by infusion of factor VIII concentrates prepared from either human plasma or by recombinant DNA technology.
Hemophilia B
Hemophilia B results from deficiencies in factor IX. The prevalence of hemophilia B is approximately one-tenth that of hemophilia A. All patients with hemophilia B have prolonged coagulation time and decreased factor IX clotting activity. Like hemophilia A, there are severe, moderate and mild forms of hemophilia B and reflect the factor IX activity in plasma.
At least 300 unique factor IX mutations have been identified, 85% are point mutations, 3% are short nucleotide deletions or insertions and 12% are gross gene alterations.
Disorders of Fibrinogen and Factor XIII
Several cardivascular risk factors are associated with abnormalities in fibrinogen. As a result of the acute-phase response or through other poorly understood mechanisms, elevated plasma fibrinogen levels have been observed in patients with coronary artery disease, diabetes, hypertension, peripheral artery disease, hyperlipoproteinemia and hypertriglyceridemia. In addition, pregnancy, menopause, hypercholesterolemia, use of oral contraceptives and smoking lead to increased plasma fibrinogen levels.
Although rare, there are inherited disorders in fibrinogen. These disorders include afibrinogenemia (a complete lack of fibrinogen), hypofibrinogenemia (reduced levels of fibrinogen) and dysfibrinogenemia (presence of dysfunctional fibrinogen). Afibrinogenemia is characterized by neonatal umbilical cord hemorrhage, ecchymoses, mucosal hemorrhage, internal hemorrhage, and recurrent abortion. The disorder is inherited in an autosomal recessive manner. Hypofibrinogenemia is characterized by fibrinogen levels below 100mg/dL (normal is 250-350mg/dL) and can be either aquired or inherited. Symptoms of hypofibrinogememia are similar to, but less severe than, afibrinogenemia. Dysfibrinogenemias are extremely heterogeneous affecting any of the functional properties of fibrinogen. Clinical consequences of dysfibrinogenemias include hemorrhage, spontaneous abortion and thromboembolism.
Factor XIII is the proenzyme form of plasma transglutaminase and is activated by thrombin in the presence of calcium ions. Active factor XIII catalyzes the cross-linking of fibrin monomers. Factor XIII is a tetramer of two two different peptides, a and b (forming a2b2). Hereditary deficiencies (autosomal recessive) occur resulting in the absence of either subunit. Clinical manifestation of factor XIII deficiency is delayed bleeding although primary hemostasis is normal. Deficiency leads to neonatal umbilical cord bleeding, intracranial hemorrhage and soft tissue hematomas.
von Willebrand Disease
von Willebrand disease (vWD) is due to inherited deficiency in von Willebrand factor (vWF). vWD is the most common inherited bleeding disorder of humans. Using sensitive laboratory testing, abnormalities in vWF can be detected in approximately 8000 people per million. Clinically significant vWD occurs in approximatley 125 people per million. This is a frequencey at least twice that of hemophilia A.
Deficiency of vWF results in defective platelet adhesion and causes a secondary deficiency in factor VIII. The result is that vWF deficiency can cause bleeding that appears similar to that caused by platelet dysfunction or hemophilia. vWD is an extremely heterogeneous disorder that has been classified into several major subtypes. Type I vWD is the most common and is inherited as an autosomal dominant trait. This variant is due to simple quantitative deficiency of all vWF multimers. Type 2 vWD is also subdivided further dependent upon whether the dysfunctional protein has decreased or paradoxically increased function in certain laboratory tests of binding to platelets. Type 3 vWD is clinically severe and is characterized by recessive inheritence and virtual absence of vWF.
Factor XI and Contact Activation
When blood makes contact with negatively charged surfaces it triggers a series of interactions that involve factor XI, prekallikrein and high molecular weight kininogen leading to blood coagulation. This process is referred to as contact activation. Deficiency in factor XI confers an injury-related bleeding tendency. This deficiency was identified in 1953 and originally termed hemophilia C. Factor XI deficiency is very common in Ashkenazic Jews and is inherited as an autosomal disorder with either homozygosity or compound heterozygosity. Three independent point mutations in factor XI have been identified.
Antithrombin Deficiency
Antithrombin functions to inhibit several activated coagulation factors including thrombin, factor IXa and factor Xa, by forming a stable complex with the various factors.. Heparin and heparan sulfates increase the activity of antithrombin at least 1000 fold.
Deficiency in antithrombin is seen in approximately 2% of patients with venous thromboembolic disease. Inheritence occurs as an autosomal dominant trait. The prevalence of symptomatic antithrombin deficiency ranges from 1 per 2000 to 1 per 5000 in the general population. Deficiencies results from mutations that affect synthesis or stability of antithrombin or from mutations that affect the protease and/or heparin binding sites of antithrombin.
Clinical manifestations of antithrombin deficiency include deep vein thrombosis and pulmonary embolism. Arterial thrombosis is rare in anththrombin deficiency. Thrombosis may occur spontaneously or in association with surgery, trauma or pregnancy. Treatment of acute episodes of thrombosis is by infusion of heparin (for 5-7 days) followed by oral anticoagulant therapy.
|
back to the top
Pharmacological Intervention in Bleeding
Coumarin drugs, such as warfarin as well as the glycosaminoglycans, heparin and heparan sulfate, are useful as anticoagulants. Heparin is useful as an anticoagulant because it binds to, and activates, antithrombin III which then inhibits the serine proteases of the coagulation cascade. Heparin is abundant in grnaules of the mast cells that line the vasculature. In response to injury, the heparin is released and inhibits coagulation. The coumarin drugs inhibit coagulation by inhibiting the vitamin K-dependent g-carboxylation reactions necessary to the function of thrombin, and factors VII, IX, and X as well as proteins C and S. These drugs act by inhibiting the reduction of the quinone derivatives of vitamin K to their active hydroquinone forms. Because of the mode of action of coumarin drugs, it takes several days for their maximum effect to be realized. For this reason, heparin is normally administered first followed by warfarin or warfarin-related drugs.
The plasminogen activators also are useful for controlling coagulation. Because tPA is highly selective for the degradation of fibrin in clots, it is extremely useful in restoring the patency of the coronary arteries following thrombosis, in particular during the short period following myocardial infarct. Streptokinase (an enzyme from the Streptococci bacterium) is another plasminogen activator useful from a therapeutic standpoint. However, it is less selective than tPA, being able to activate circulating plasminogen as well as that bound to a fibrin clot.
Aspirin is an important inhibitor of platelet activation. By virtue of inhibiting the activity of cyclooxygenase, aspirin reduces the production of TXA2. Aspirin also reduces endothelial cell production of prostacyclin (PGI2), an inhibitor of platelet aggregation and a vasodilator. Since endothelial cells regenerate active cyclooxygenase faster than platelets, the net effect of aspirin is more in favor of endothelial cell-mediated inhibition of the coagulation cascade.
back to the top
Back to Topics<<<<
This article has been modified by Dr. M. Javed Abbas.
If you have any comments please do not hesitate to sign my Guest Book.
20:52 21/12/2002
|