Introduction
Although the two most prominent fates of amino acids are incorporation into protein and oxidative degradation, the biosynthesis of numerous physiologically important compounds require the availability of amino acids and their constituent nitrogen.
Aside from its importance as the prosthetic group of hemoglobin and a small number of enzymes (e.g., redox cytochromes and the P450 class of detoxifying cytochromes), heme is important because a number of genetic disease states are associated with deficiencies of the enzymes used in its biosynthesis. Some of these disorders are readily diagnosed because they cause d-aminolevulinic acid, (ALA) and other abnormally colored heme intermediates to appear in the circulation, the urine, and in other tissues such as teeth and bones. Some disorders of heme biosynthesis are more insidious such as the various porphyrias, a list of which can be found below and in the Inborn Errors Page.
back to the top
Synthesis of Porphobilinogen and Heme
The first reaction in heme biosynthesis takes place in the mitochondrion and involves the condensation of 1 glycine and 1 succinylCoA by the pyridoxal phosphate-containing enzyme, d-aminolevulinic acid synthase (ALA synthase). This reaction is both the rate-limiting reaction of heme biosynthesis, and the most highly regulated reaction (see Regulation below). Mitochondrial d-aminolevulinic acid (ALA) is transported to the cytosol, where ALA dehydratase (also called porphobilinogen synthase or hydroxymethylbilane synthase) dimerizes 2 molecules of ALA to produce the pyrrole ring compound porphobilinogen.
The next step in the pathway involves the head-to-tail condensation of 4 molecules of porphobilinogen to produce the linear tetrapyrrole intermediate, hydroxymethylbilane. The enzyme for this condensation is porphobilinogen deaminase (PBG deaminase). This enzyme is also called uroporphyrinogen I synthase. Hydroxymethylbilane has two main fates. The most important is regulated, enzymatic conversion to uroporphyrinogen III, the next intermediate on the path to heme. This step is mediated by a holoenzyme comprised of uroporphyrinogen synthase plus a protein known as uroporphyrinogen III cosynthase.
Genetic defects that cause increased ALA synthase activity or decreased uroporphyrinogen I synthase activity lead to the disease known as acute intermittent porphyria, which is diagnosed by the excretion of excess porphobilinogen (a condition that is not obvious from the color of the urine).
Genetic defects in enzymes beyond hydroxymethylbilane in the biosynthetic pathway result in the accumulation of excess hydroxymethylbilane, leading to an alternative non-enzymatic cyclization. In erythropoietic uprotoporphyria, bone marrow uroporphyrinogen III cosynthase is only present at about 30% of the normal level; the result is that massive amounts of Type I uroporphyrinogen and its highly colored oxidation products are found in the urine and deposited in a variety of tissues, including teeth and bones.
In the cytosol, the acetate substituents of uroporphyrinogen (normal uroporphyrinogen III or abnormal uroporphyrinogen I) are all decarboxylated by the enzyme uroporphyrinogen decarboxylase. The resultant products have methy groups in place of acetate and are known as coproporphyrinogens, with coproporphyrinogen III being the , important, normal intermediate in heme synthesis.
Coproporphyrinogen III is transported to the interior of the mitochondrion, where 2 propionate residues are decarboxylated, yielding vinyl substituents on the 2 pyrrole rings. The colorless product is protoporphyrinogen IX. In the mitochondrion, protoporphyrinogen IX is converted to protoporphyrin IX (structure shown below) by protoporphyrinogen IX oxidase. The oxidase reaction requires molecular oxygen and results in the loss of 6 protons and 6 electrons, yielding a completely conjugated ring system, which is responsible for the characteristic red color to hemes. The final reaction in heme synthesis also takes place in the mitochondrion and involves the insertion of the iron atom into the ring system generating heme b. The enzyme catalyzing this reaction is known as ferrochelatase.
 |
| Protoporphyrin IX |
The enzymes ferrochelatase, ALA synthase and ALA dehydratase (a sulfhydryl containing enzyme) are highly sensitive to inhibition by heavy metal poisoning. Indeed, a characteristic of lead poisoning is an increase in ALA in the circulation in the absence of an increase in porphobilinogen.
back to the top
Regulation of Heme Biosynthesis
Although heme is synthesized in virtually all tissues, the principal sites of synthesis are erythroid cells (~85%) and hepatocytes (accounting for nearly all the rest of heme synthesis). The differences in these two tissues and their needs for heme result in quite different mechanisms for regulation of heme biosynthesis.
In hepatocytes, heme is required for incorporation into the cytochromes, in particular, the P450 class of cytochromes that are important for detoxification. In addition numerous cytochromes of the oxidative-phosphorylation pathway contain heme. The rate-limiting step in hepatic heme biosynthesis occurs at the ALA synthase catalyzed step, which is the committed step in heme synthesis. The Fe3+ oxidation product of heme is termed hemin. Hemin acts as a feed-back inhibitor on ALA synthase. Hemin also inhibits transport of ALA synthase from the cytosol (its' site of synthesis) into the mitochondria (its' site of action) as well as represses synthesis of the enzyme.
In erythroid cells all of the heme is synthesized for incorporation into hemoglobin and occurs only upon differentiation when synthesis of hemoglobin proceeds. When red cells mature both heme and hemoglobin synthesis ceases. The heme (and hemoglobin) must, therefore, survive for the life of the erythrocyte (normally this is 120 days). In reticulocytes (immature erythrocytes) heme stimulates protein synthesis. The mechanism of this mode of heme-mediate regulation of protein synthesis is described in the Protein Synthesis section. Additionally, control of heme biosynthesis in erythrocytes occurs at numerous sites other than at the level of ALA synthase. Control has been shown to be exerted on ferrochelatase, the enzyme responsible for iron insertion into protoporphyrin IX, and on porphobilinogen deaminase.
back to the top
Heme Metabolism
The largest repository of heme in the human body is in red blood cells, which have a life span of about 120 days. There is thus a turnover of about 6 g/day of hemoglobin, which presents 2 problems. First, the porphyrin ring is hydrophobic and must be solubilized to be excreted. Second, iron must be conserved for new heme synthesis.
Normally, senescent red blood cells and heme from other sources are engulfed by cells of the reticuloendothelial system. The globin is recycled or converted into amino acids, which in turn are recycled or catabolized as required. Heme is oxidized, with the heme ring being opened by the endoplasmic reticulum enzyme, heme oxygenase. The oxidation step requires heme as a substrate, and any hemin (Fe3+) is reduced to heme (Fe2+) prior to oxidation by heme oxygenase. The oxidation occurs on a specific carbon producing the linear tetrapyrrole biliverdin, ferric iron (Fe3+), and carbon monoxide (CO). This is the only reaction in the body that is known to produce CO. Most of the CO is excreted through the lungs, with the result that the CO content of expired air is a direct measure of the activity of heme oxygenase in an individual.
In the next reaction a second bridging methylene (between rings III and IV) is reduced by biliverdin reductase, producing bilirubin. Bilirubin is significantly less extensively conjugated than biliverdin causing a change in the color of the molecule from blue-green (biliverdin) to yellow-red (bilirubin). The latter catabolic changes in the structure of tetrapyrroles are responsible for the progressive changes in color of a hematoma, or bruise, in which the damaged tissue changes its color from an initial dark blue to a red-yellow and finally to a yellow color before all the pigment is transported out of the affected tissue. Peripherally arising bilirubin is transported to the liver in association with albumin, where the remaining catabolic reactions take place.
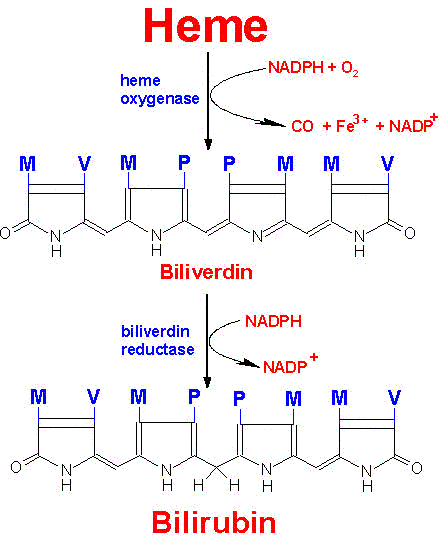 |
Pathway for the degradation of heme to bilirubin
Substituents: M=methyl, P=proprionic, V=vinyl |
In hepatocytes, UDP glucuronyl transferase adds 2 equivalents of glucuronic acid to bilirubin to produce the more water soluble, bilirubin diglucuronide derivative. The increased water solubility of the tetrapyrrole facilitates its excretion with the remainder of the bile as the bile pigments.
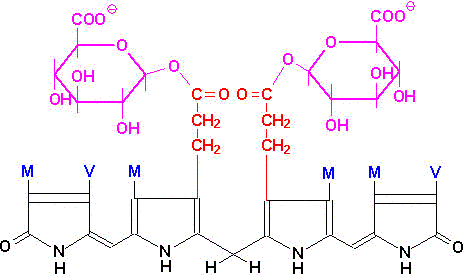 |
| Bilirubin diglucuronide |
In individuals with abnormally high red cell lysis, or liver damage with obstruction of the bile duct, the bilirubin and its precursors accumulate in the circulation; the result is hyperbilirubinemia, the cause of the abnormal body pigmentation known as jaundice. In normal individuals, intestinal bilirubin is acted on by bacteria to produce the final porphyrin products, urobilinogens and urobilins, that are found in the feces. Bilirubin and its catabolic products are collectively known as the bile pigments.
back to the top
Clinical Aspect of Heme Metabolism
Clinical problems associated with heme metabolism are of two types. Disorders that arise from defects in the enzymes of heme biosynthesis are termed the porphyrias and cause elevations in the serum and urine content of intermediates in heme synthesis. Inherited disorders in bilirubin metabolism lead to hyperbilirubinemia.
Bilirubin is potentially toxic waste product of heme catabolism. The body eliminates bilirubin by transporting it to the liver bound to albumin in the serum. In the liver it is conjugated with glucuronate which renders it water soluble. The glucuronide conjugate is then excreted in the bile. Persons with extreme elevation in unconjugated bilirubin are susceptible to bilirubin encephalopathy, also referred to as kernicterus. Accumulation of bilirubin in the plasma and tissues results in jaundice.
Several inherited disorders in bilirubin metabolism have been identified. Gilbert syndrome and Crigler-Najjar syndrome result from predominantly unconjugated hyperbilirubinemia. Dubin-Johnson syndrome and Rotor syndrome result from conjugated hyperbilirubinemia.
The porphyrias are both inherited and acquired disorders in heme synthesis. These disorders are classified as either erythroid or hepatic, depending upon the principal site of expression of the enzyme defect. Eight different porphyrias have been classified. With the exception of the reaction catalyzed by ALA synthase, defects in each of the other enzymes of heme synthesis have been identified. The most commonly occuring porphyria is acute intermittent porphyria, AIP which is caused by a defect in porphobilinogen deaminase, (PBG deaminase). This enzyme is also called hydroxymethylbilane synthase or uroporphyrinogen synthase.
All of the porphyrias lead to excretion of heme biosynthetic byproducts that turn the urine red and when deposited in the teeth turn them reddish brown. Accumulation of these byproducts in the skin renders it extremely sensitive to sunlight causing ulceration and disfiguring scars. Increased hair growth is also a symptom of the porphryias leading to appearance of fine hairs over the entire face and on the extremeties.
| Porphyria | Enzyme Defect | Primary Symptom |
| Erythropoietic Class |
| Congenital erythropoietic porphyria, CEP | Uroporphyrinogen III cosynthase | Photosensitivity |
| Erythropoietic protoporphyria, EPP | Ferrochelatase | Photosensitivity |
| Hepatic Class |
| ALA dehydratase deficiency porphyria, ADP | ALA dehydratase | Neurovisceral |
| Acute intermittent porphyria, AIP | PBG deaminase | Neurovisceral |
| Hereditary coproporphyria, HCP | Coproporphyrinogen oxidase | Neurovisceral, some photosensitivity |
| Variegate porphyria, VP | Protoporphyrinogen oxidase | Neurovisceral, some photosensitivity |
| Porphyria cutanea tarda, PCT | Uroporphyrinogen decarboxylase | Photosensitivity |
| Hepatoerythropoietic porphyria, HEP | Uroporphyrinogen decarboxylase | Photosensitivity, some neurovisceral |
back to the top
Back to Topics<<<<
This article has been modified by Dr. M. Javed Abbas.
If you have any comments please do not hesitate to sign my Guest Book.
20:48 21/12/2002
|