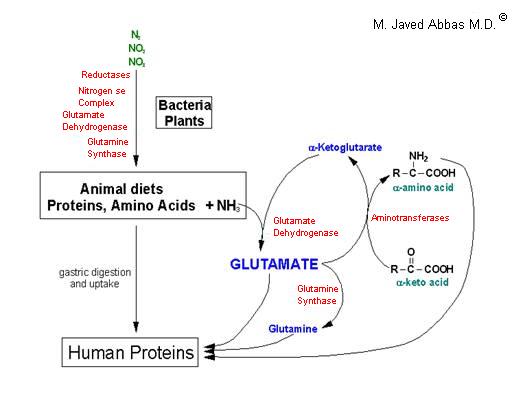
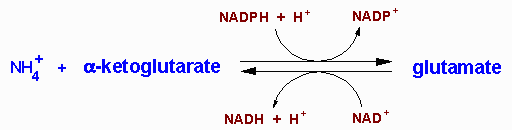
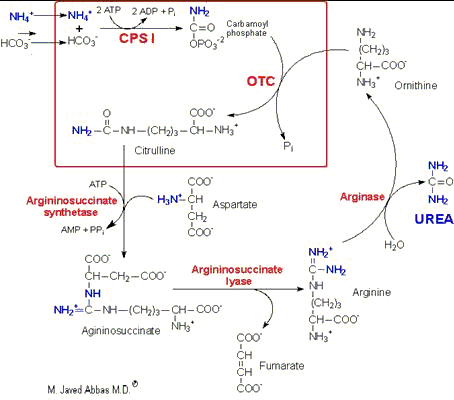
| UCD | Enzyme Deficiency | Symptoms/Comments |
| Type I Hyperammonemia | Carbamoylphosphate synthetase I | with 24h - 72h after birth infant becomes lethargic, needs stimulation to feed, vomiting, increasing lethargy, hypothermia and hyperventilation; without measurement of serum ammonia levels and appropriate intervention infant will die: treament with arginine which activates N-acetylglutamate synthetase |
| N-acetylglutamate synthetase Deficiency | N-acetylglutamate synthetase | severe hyperammonemia, mild hyperammonemia associated with deep coma, acidosis, recurrent diarrhea, ataxia, hypoglycemia, hyperornithinemia: treatment includes administration of carbamoyl glutamate to activate CPS I |
| Type 2 Hyperammonemia | Ornithine transcarbamoylase | most commonly occurring UCD, only X-linked UCD, ammonia and amino acids elevated in serum, increased serum orotic acid due to mitochondrial carbamoylphosphate entering cytosol and being incorporated into pyrimidine nucleotides which leads to excess production and consequently excess catabolic products: treat with high carbohydrate, low protein diet, ammonia detoxification with sodium phenylacetate or sodium benzoate |
| Classic Citrullinemia | Argininosuccinate synthetase | episodic hyperammonemia, vomiting, lethargy, ataxia, siezures, eventual coma: treat with arginine administration to enhance citrulline excretion, also with sodium benzoate for ammonia detoxification |
| Argininosuccinic aciduria | Argininosuccinate lyase
(argininosuccinase) | episodic symptoms similar to classic citrullinemia, elevated plasma and cerebral spinal fluid argininosuccinate: treat with arginine and sodium benzoate |
| Hyperargininemia | Arginase | rare UCD, progressive spastic quadriplegia and mental retardation, ammonia and arginine high in cerebral spinal fluid and serum, arginine, lysine and ornithine high in urine: treatment includes diet of essential amino acids excluding arginine, low protein diet |