Actions of Insulin
The major function of insulin is to counter the concerted action of a number of hyperglycemia-generating hormones and to maintain low blood glucose levels. Because there are numerous hyperglycemic hormones, untreated disorders associated with insulin generally lead to severe hyperglycemia and shortened life span.
Insulin is synthesized as a preprohormone in the b cells of the islets of Langerhans. Its signal peptide is removed in the cisternae of the endoplasmic reticulum and it is packaged into secretory vesicles in the Golgi, folded to its native structure, and locked in this conformation by the formation of 2 disulfide bonds. Specific protease activity cleaves the center third of the molecule, which dissociates as C peptide, leaving the amino terminal B peptide disulfide bonded to the carboxy terminal A peptide.
Insulin secretion from b cells is principally regulated by plasma glucose levels, but the precise mechanism by which the glucose signal is transduced remains unclear. One possibility is that the increased uptake of glucose by pancreatic b-cells leads to a concommitant increase in metabolism. The increase in metabolism leads to an elevation in the ATP/ADP ratio. This in turn leads to an inhibition of an ATP-sensitive K+ channel. The net result is a depolarization of the cell leading to Ca2+ influx and insulin secretion.
Chronic increases in numerous other hormones---including growth hormone, placental lactogen, estrogens, and progestins --- up-regulate insulin secretion, probably by increasing the preproinsulin mRNA and enzymes involved in processing the increased preprohormone.
In contrast, epinephrine diminishes insulin secretion by a cAMP-coupled regulatory path. In addition, epinephrine counters the effect of insulin in liver and peripheral tissue, where it binds to b-adrenergic receptors, induces adenylate cycles activity, increases cAMP, and activates PKA similarly to that of glucagon. The latter events induce glycogenolysis and gluconeogenesis, both of which are hyperglycemic and which thus counter insulin's effect on blood glucose levels. In addition, epinephrine influences glucose homeostasis through interaction with a-adrenergic receptors.
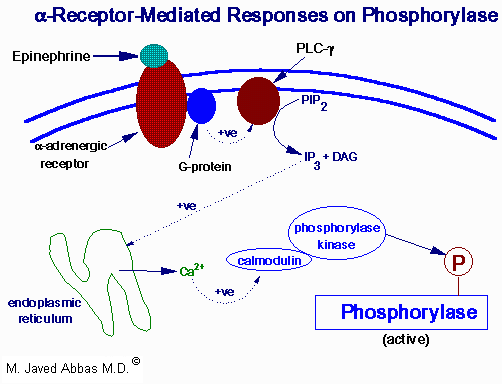 |
| Pathways involved in the regulation of glycogen phosphorylase by epinephrine activation of a-adrenergic receptors. See Glycogen Metabolism for details of the epinephrine action. PLC-g is phospholipase C-g. The substrate for PLC-g is phosphatidylinositol-4,5-bisphosphate (PIP2) and the products are IP3, inositol trisphosphate and DAG, diacylglycerol. Similar calmodulin-mediated phosphorylations lead to inhibition of glycogen synthase.
|
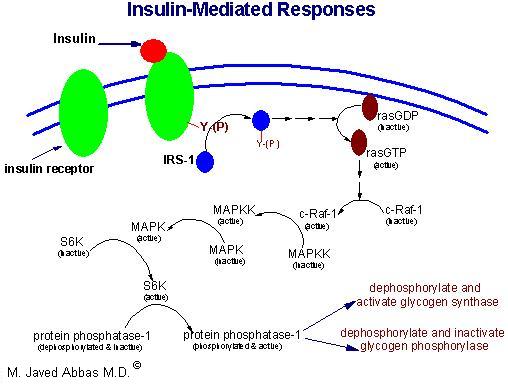
Insulin secreted by the pancreas is directly infused via the portal vein to the liver, where it exerts profound metabolic effects. These effects are the response of the activation of the insulin receptor which belongs to the class of cell surface receptors that exhibit intrinsic tyrosine kinase activity (see Signal Transduction). With respect to hepatic glucose homeostasis, the effects of insulin receptor activation are specific phosphorylation events that lead to an increase in the storage of glucose with a concomitant decrease in hepatic glucose release to the circulation (see below).
In most other tissues insulin increases the number of plasma membrane glucose transporters, but in liver glucose uptake is dramatically increased because of increased activity of the enzymes glucokinase, phosphofructokinase-1 (PFK-1), and pyruvate kinase (PK), the key regulatory enzymes of glycolysis. The latter effects are induced by insulin-dependent activation of phosphodiesterase, with decreased PKA activity and diminished phosphorylation of pyruvate kinase and phosphofructokinase-2, PFK-2. Dephosphorylation of pyruvate kinase increases its' activity while dephosphorylation of PFK-2 renders it active as a kinase. The kinase activity of PFK-2 converts fructose-6-phosphate into fructose-2,6-bisphosphate (F2,6BP). F2,6BP is a potent allosteric activator of the rate limiting enzyme of glycolysis, PFK-1, and an inhibitor of the gluconeogenic enzyme, fructose-1,6-bisphosphatase. In addition, phophatases specific for the phosphorylated forms of the glycolytic enzymes increase in activity under the influence of insulin. All these events lead to conversion of the glycolytic enzymes to their active forms and consequently a significant increase in glycolysis. In addition, glucose-6-phosphatase activity is down-regulated. The net effect is an increase in the content of hepatocyte glucose and its phosphorylated derivatives, with diminished blood glucose.
In addition to the latter events, diminished cAMP and elevated phosphatase activity combine to convert glycogen phosphorylase to its inactive form and glycogen synthase to its active form, with the result that not only is glucose funneled to glycolytic products, but glycogen content is increased as well.
Insulin generates its intracellular effects by binding to a plasma membrane receptor, which is the same in all cells. The receptor is a disulfide-bonded glycoprotein. One function of insulin (aside from its role in signal transduction) is to increase glucose transport in extrahepatic tissue is by increasing the number of glucose transport molecules in the plasma membrane. Glucose transporters are in a continuous state of turnover. Increases in the plasma membrane content of transporters stem from an increase in the rate of recruitment of new transporters into the plasma membrane, deriving from a special pool of preformed transporters localized in the cytoplasm.
In addition to its role in regulating glucose metabolism, insulin stimulates lipogenesis, diminishes lipolysis, and increases amino acid transport into cells. Insulin also modulates transcription, altering the cell content of numerous mRNAs. It stimulates growth, DNA synthesis, and cell replication, effects that it holds in common with the IGFs and relaxin.
back to the top
Definition of Diabetes
Diabetes is any disorder characterized
by excessive urine excretion. The most common form of diabetes is diabetes
mellitus, a metabolic disorder in which
there is an inability to oxidize carbohydrate due to disturbances in insulin
function. Diabetes mellitus is characterized by elevated glucose in the
plasma and episodic ketoacidosis. Additional symptoms of diabetes mellitus
include excessive thirst, glucosuria, polyuria, lipemia and hunger. If
left untreated the disease can lead to fatal ketoacidosis. Other forms
of diabetes include diabetes insipidus
and brittle diabetes. Diabetes
insipidus is the result of a deficiency of antidiuretic
hormone. The major symptom of diabetes
insipidus (excessive urine output) results from an inability of the kidneys
to resorb water. Brittle diabetes is a form that is very difficult to control.
It is characterized by unexplained oscillations between hypoglycemia and
acidosis.
Criteria, which clinically establish
an individual as suffering from diabetes mellitus, include:
- 1. Having a fasting plasma
glucose level in excess of 140mg/dL.
- 2. or having plasma glucose
levels in excess of 200mg/dL at two times points during a glucose
tolerance test (GTT), one of which must be within 2 hrs of ingestion
of glucose.
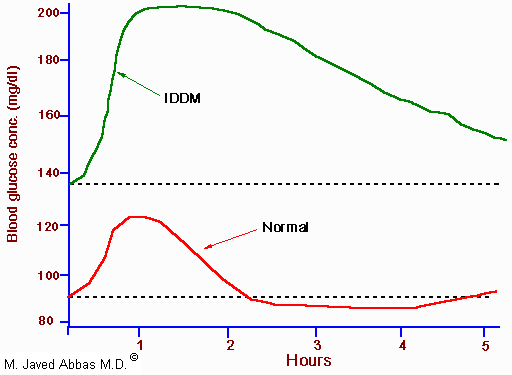 |
| Glucose tolerance curve for a normal person and one with insulin-dependent diabetes mellitus (IDDM, Type 1 diabetes). The dotted lines indicate the range of glucose concentration expected in a normal individual. |
back to the top
Types of Diabetes Mellitus
Diabetes mellitus is a heterogeneous clinical disorder
with numerous causes. Two main classifications of diabetes mellitus exist,
idiopathic and secondary.
Idiopathic diabetes is divided into two main types; insulin dependent and non-insulin-depenedent. Insulin-dependent diabetes mellitus (IDDM, also called Type 1 diabetes) is defined by the development of ketoacidosis in the absence of insulin therapy. Type 1 diabetes most often manifests in childhood (hence also called juvenile onset diabetes) and is the result of an autoimmune destruction of the b-cells of the pancreas. Non-insulin-dependent diabetes mellitus (NIDDM or Type 2 diabetes) is characterized by milder hyperglycemia and rarely leads to ketoacidosis. Type 2 diabetes generally manifests after age 40 and is therefore has the obselete name of adult onset-type diabetes. Type 2 diabetes results from genetics defects that cause both insulin resistance and insulin deficiency. There are two main forms of type 2 diabetes:
- 1. Late onset associated with obesity.
- 2. Late onset not associated with obesity.
Secondary, or other specific types of diabetes mellitus are the result of many causes
including:
- 1. Maturity onset type diabetes of the young (MODY) was previously considered to be a third form of type 2 diabetes. However, with the discovery of specific mutations leading to MODY, it is now classified under secondary or other specific types of diabetes. MODY is characterized by onset prior to age 25. All cases to date have shown impaired b-cell function. Patients may also exhibit insulin resistance and late b-cell failure. Mutations in 5 genes have been linked to MODY: MODY-1 results from a defect in the hepatic nuclear factor-4a (HNF-4a) gene; MODY-2 is due to a defect in the glucokinase gene; MODY-3 is caused by defects in the HNF-1a gene; MODY-4 is due to a mutation in the insulin promoter factor-1 (IPF-1) gene; and finally defects in HNF-1b are a rare cause of MODY.
- 2. Pancreatic disease: Pancreatectomy leads to the clearest example of secondary diabetes. Cystic fibrosis and pancreatitis can also lead to destruction of the pancreas.
- 3. Endocrine disease: Some tumors can produce counter-regulatory hormones that oppose the action of insulin or inhibit insulin secretion. These counter-regulatory hormones are glucagon, epinephrine, growth hormone and cortisol.
- a. Glucagonomas are pancreatic cancers that secrete glucagon.
- b. Pheochromocytomas secrete epinephrine.
- c. Cushing syndrome results in excess cortisol secretion.
- d. Acromegaly results in excess growth hormone production.
- 4. Drug-induced diabetes; treatment with glucocorticoids and diuretics can interfere with insulin function.
- 5. Anti-insulin receptor autoantibodies (Type
B insulin resistance)
- 6. Mutations in the insulin gene
- 7. Mutations
in insulin receptor gene which lead to the syndromes listed below. Two clinical features are common in all syndromes that result from mutations in the insulin receptor gene: acanthosis nigricans and hyperandrogenism (the latter being observed only in females).
- a. Leprachaunism
- b. Rabson-Mendenhall syndrome
- c. Type A insulin resistance
- 8. Gestational diabetes; this syndrome sets in during pregnancy
and usually resolves itself following childbirth.
- 9. Many other genetic syndromes have either diabetes or impaired glucose tolerance associated with them; lipoatrophic diabetes, Wolfram syndrome, Down syndrome, Klinefelter syndrome (XXY males), Turner syndrome, myotonic dystrophy, muscular dystrophy, Huntington disease, Friedrich ataxia (associated with deficiency in purine nucleotide phosphorylase), Prader-Willi syndrome, Werner syndrome, Cockayne syndrome, and others such as those indicated above in 2 and 6.
back to the top
Insulin-Dependent Diabetes Mellitus (IDDM)
Etiology of IDDM
Type 1 diabetes has been shown to be the result of an autoimmune reaction to antigens of the islet cells of the pancreas. There is a strong association between IDDM and other endocrine autoimmunities (e.g. Addison disease). Additionally, there is an increased prevalence of autoimmune disease in family members of IDDM patients.
Types of Autoantibodies:
- 1. Islet cell cytoplasmic antibodies: The primary antibodies found in 90% of type 1 diabetics are against islet cell cytoplasmic proteins (termed ICCA, islet cell cytoplasmic antibodies). In non-diabetics ICCA frequency is only 0.5% - 4%. The presence of ICCA is a highly accurate predictor of future development of IDDM.
- ICCA are not specific for the b-cells and recognize antigens in other cell types in the islet. However, the autoimmune attack appears to selectively destroy b-cells. Therefore, the antibodies may play a primary role in the destruction of islet cells. It is an equally likely possibility that the production of anti-islet antibodies occurs as a result of the destruction of b-cells. Whether a direct cause or an effect of islet cell destruction, the titer of the ICCA tends to decline over time.
- 2. Islet cell surface antibodies: Autoantibodies directed against cell-surface antigens (ICSA) have also been described in as many as 80% of type 1 diabetics. Similar to ICCA, the titer of ICSA declines over time. Some patients with type 2 diabetes have been identified that are ICSA positive.
- 3. Specific antigenic targets of islet cells: Antibodies to glutamic acid decarboxylase (GAD) have been identified in over 80% of patients newly diagnosed with IDDM. Like ICCA, anti-GAD antibodies decline over time in type 1 diabetics. The presence of anti-GAD antibodies is a strog predictor of the future development of IDDM in high-risk populations. Anti-insulin antibodies (IAA) have been identified in IDDM patients and in relatives at risk to develop IDDM. These IAA are detectable even before the onset of insulin therapy in type 1 diabetics. IAA are detectable in around 40% of young children with IDDM.
Pathophysiology of IDDM
The autoimmune destruction of pancreatic b-cells leads to a deficiency of insulin secretion. It is this loss of insulin secretion that leads to the metabolic derangements associated with IDDM. In addition to the loss of insulin secretion, the function of pancreatic a-cells is also abnormal. There is excessive secretion of glucagon in IDDM patients. Normally, hyperglycemia leads to reduced glucagon secretion. However, in patients with IDDM, glucagon secretion is not suppressed by hyperglycemia. The resultant inappropriately elevated glucagon levels exacerbates the metabolic defects due to insulin deficiency (see below). The most pronounced example of this metabolic disruption is that patients with IDDM rapidly develop diabetic ketoacidosis in the absence of insulin administration. If somatostatin is administered to suppress glucagon secretion, there is a concommitant suppression in the rise of glucose and ketone bodies. Particularly problematic for long term IDDM patients is an impaired ability to secrete glucagon in response to hypoglycemia. This leads to potentially fatal hypoglycemia in response to insulin treatment in these patients.
Although insulin deficiency is the primary defect in IDDM, in patients with poorly controlled IDDM there is also a defect in the ability of target tissues to respond to the administration of insulin. There are multiple biochemical mechanisms that account for this impairment of tissues to respond to insulin. Deficiency in insulin leads to elevated levels of free fatty acids in the plasma as a result of uncontrolled lipolysis in adipose tissue. Free fatty acids suppress glucose metabolism in peripheral tissues such as skeletal muscle. This impairs the action of insulin in these tissues, i.e. the promotion of glucose utilization. Additionally, insulin deficiency decreases the expression of a number of genes necessary for target tissues to respond normally to insulin -- such as glucokinase in liver and the GLUT 4 class of glucose transporters in adipose tissue.
The major metabolic derangements which result from insulin deficiency in IDDM are impaired glucose, lipid and protein metabolism.
Glucose Metabolism: Uncontrolled IDDM leads to increased hepatic glucose output. First, liver glycogen stores are mobilized then hepatic gluconeogenesis is used to produce glucose. Insulin deficiency also impairs non-hepatic tissue utilization of glucose. In particular in adipose tissue and skeletal muscle, insulin stimulates glucose uptake. This is accomplished by insulin-mediated movement of glucose transporter proteins to the plasma membrane of these tissues. Reduced glucose uptake by peripheral tissues in turn leads to a reduced rate of glucose metabolism. In addition, the level of hepatic glucokinase is regulated by insulin. Therefore, a reduced rate of glucose phosphorylation in hepatocytes leads to increased delivery to the blood. Other enzymes involved in anabolic metabolism of glucose are affected by insulin (primarily through covalent modifications). The combination of increased hepatic glucose production and reduced peripheral tissues metabolism leads to elevated plasma glucose levels. When the capacity of the kidneys to absorb glucose is surpassed, glucosuria ensues. Glucose is an osmotic diuretic and an increase in renal loss of glucose is accompanied by loss of water and electrolytes, termed polyuria. The result of the loss of water (and overall volume) leads to the activation of the thirst mechanism (polydipsia). The negative caloric balance which results from the glucosuria and tissue catabolism leads to an increase in appetite and food intake (polyphagia).
Lipid Metabolism: One major role of insulin is to stimulate the storage of food energy following the consumption of a meal. This energy storage is in the form of glycogen in hepatocytes and skeletal muscle. Additionally, insulin stimulates hepatocytes to synthesize triglycerides and storage of triglycerides in adipose tissue. In opposition to increased adipocyte storage of triglycerides is insulin-mediated inhibition of lipolysis. In uncontrolled IDDM there is a rapid mobilization of triglycerides leading to increased levels of plasma free fatty acids. The free fatty acids are taken up by numerous tissues (however, not the brain) and metablized to provide energy. Free fatty acids are also taken up by the liver.
Normally, the levels of malonyl-CoA are high in the presence of insulin. These high levels of malonyl-CoA inhibit carnitine palmitoyltransferase I, the enzyme required for the transport of fatty acyl-CoA's into the mitochondria where they are subject to oxidation for energy production. Thus, in the absence of insulin, malonyl-CoA levels fall and transport of fatty acyl-CoA's into the mitochondria increases. Mitochondrial oxidation of fatty acids generates acetyl-CoA which can be further oxidized in the TCA cycle. However, in hepatocytes the majority of the acetyl-CoA is not oxidized by the TCA cycle but is metabolized into the ketone bodies, acetoacetate and b-hydroxybutyrate. These ketone bodies leave the liver and are used for energy production by the brain, heart and skeletal muscle. In IDDM, the increased availability of free fatty acids and ketone bodies exacerbates the reduced utilization of glucose furthering the ensuing hyperglycemia. Production of ketone bodies in excess of the bodies ability to utilize them leads to ketoacidosis. In diabetics, this can be easily diagnosed by smelling the breath. A spontaneous breakdown product of acetoacetate in acetone which is volatilized by the lungs producing a distinctive odor.
Normally, plasma triglycerides are acted upon by lipoprotein lipase (LPL), an enzyme on the surface of the endothelial cells lining the vessels. In particular, LPL activity allows fatty acids to be taken from circulating triglycerides for storage in adipocytes. The activity of LPL requires insulin and in its absence a hypertriglyceridemia results.
Protein Metabolism: Insulin regulates the synthesis of many genes, either positively or negatively that then affect overall metabolism. Insulin has a global effect on protein metabolism -- increasing the rate of protein synthesis and decreasing the rate of protein degradation. Thus, insulin deficiency will lead to increased catabolism of protein. The increased rate of proteolysis leads to elevated concentrations in plasma amino acids. These amino acids serve as precursors for hepatic and renal gluconeogensis. In liver, the increased gluconeogenesis further contributes to the hyperglycemia seen in IDDM.
back to the top
Non-Insulin-Dependent Diabetes Mellitus (NIDDM)
Etiology of NIDDM
NIDDM is characterized by a lack of the need for insulin to prevent ketoacidosis. Type 2 diabetes refers to the common form of idiopathic NIDDM. NIDDM is not an autoimmune disorder, however, there is a strong genetic correlation to the susceptibility to NIDDM. The susceptibility genes that predispose one to NIDDM have not been identified in most patients. This is due in part to the heterogeneity of the genes responsible for the susceptibility to NIDDM. Obesity is a major risk factor that predisposes one to NIDDM. Genetic studies in mice and rats have demonstrated a link between genes responsible for obesity and those that cause diabetes mellitus.
Pathophysiology of NIDDM
Unlike patients with IDDM, those with NIDDM have detectable levels of circulating insulin. On the basis of oral glucose tolerance testing the essential elements of NIDDM can be divided into 4 distinct groups; those with normal glucose tolerance, chemical diabetes (called impaired glucose tolerance), diabetes with minimal fasting hyperglycemia (fasting plasma glucose <140 mg/dL), and diabetes mellitus in association with overt fasting hyperglycemia (fasting plasma glucose >140 mg/dL). In patients with the highest levels of plasma insulin (impaired glucose tolerance group) there was also elevated plasma glucose. This indicates that these individuals are resistant to the action of insulin. In the progression from impaired glucose tolerance to diabetes mellitus the level of insulin declines indicating that patients with NIDDM have decreased insulin secretion.
Additional studies have subsequently demonstrated that both insulin resistance and insulin deficiency is common in the average NIDDM patient. Many experts conclude that insulin resistance is the primary cause of NIDDM, however, others contend that insulin deficiency is the primary cause because a moderate degree of insulin resistance is not sufficient to cause NIDDM. As indicated above, most patients with the common form of NIDDM have both defects.
A number of candidate genes have been screened for having causative roles in type 2 diabetes although none has bee shown as a significant cause of the disease. Several of these genes include glucokinase, GLUT-2 (glucose transporter), glucagon receptor, glucagon-like protein-1 (GLIP-1), glucokinase regulatory protein and hexokinase-1.
Recent evidence has demonstrated a role for a member of the nuclear hormone receptor superfamily of proteins in the etiology of type 2 diabetes. A relatively new class of drugs used to increase the sensitivity of the body to insulin are the thiazolidinedione drugs. These compounds bind to and alter the function of the peroxisome proliferator-activated receptor g (PPARg). PPARg is also a transcription factor and, when activated, binds to another transcription factor known as the retinoid X receptor (RXR). When these two proteins are complexed a specific set of genes becomes activated. PPARg is a key regulator of adipocyte differentiation, it can induce the differentiation of fibroblasts or other undifferentiated cells into mature fat cells. PPARg is also involved in the synthesis of biologically active compounds from vascular endothelial cells and immune cells.
Mutations in the gene for PPARg have been correlated with insulin resistance. It is still not completely clear how impaired PPARg signaling can affect the sensitivity of the body to insulin or indeed if the observed mutations are a direct or indirect cause of the symptoms of insulin resistance.
back to the top
Back to Topics<<<<
This article has been modified by Dr. M. Javed Abbas.
If you have any comments please do not hesitate to sign my Guest Book.
20:46 21/12/2002
|