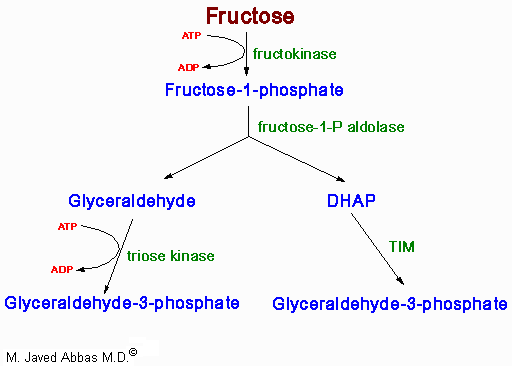
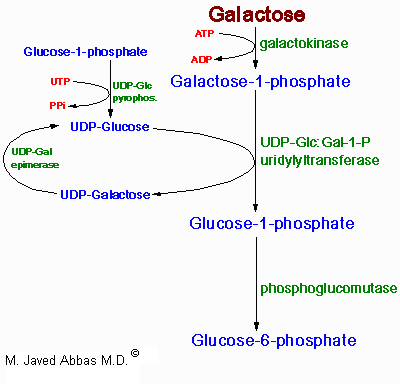
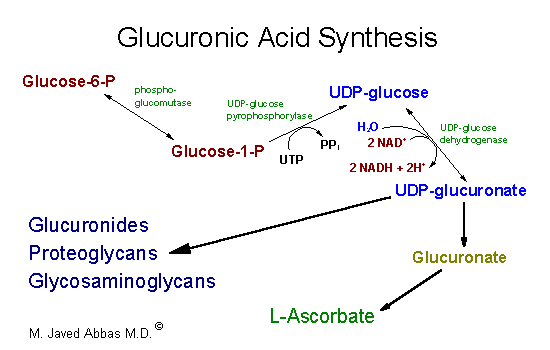
| The uronic acid pathway is
utilized to synthesize UDP-glucuronate, glucuronate and L-ascorbate. The
pathway involves the oxidation of glucosae-6-phosphate to UDP-glucuronate.
The oxidation is uncoupled from energy production. UDP-glucuronate is used
in the synthesis of glycosaminoglycan
and proteoglycans as well as forming complexes with bilirubin,
steroids and certain drugs. The glucuronate complexes form to solubilize
compounds for excretion. The synthesis of ascorbate (vitamin C)
does not occur in primates.
|