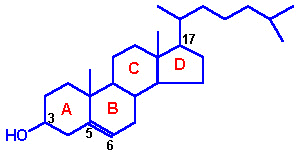
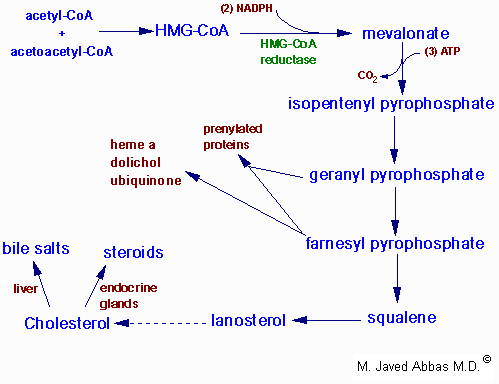
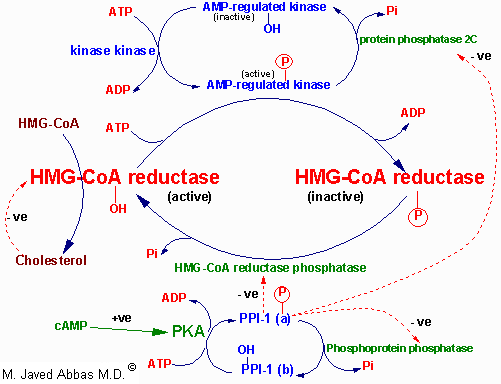
| Regulation of HMG-CoA reductase by covalent modification. HMG-CoA reductase is most active in the dephosphorylated state. Phosphorylation is catalyzed by AMP-regulated protein kinase, AMPRK, (used to be termed reductase kinase), an enzyme whose activity is also regulated by phosphorylation. Phosphorylation of AMPRK is catalyzed by kinase kinase. Hormones such as glucagon and epinephrine negatively affect cholesterol biosynthesis by increasing the activity of the inhibitor of phosphoprotein phosphatase inhibitor-1, PPI-1. Conversely, insulin stimulates the removal of phosphates and, thereby, activates HMG-CoA reductase activity. Additional regulation of HMG-CoA reductase occurs through an inhibition of its' activity as well as of its' synthesis by elevation in intracellular cholesterol levels. The mechanism of this cholesterol induced inhibition is not fully understood.
|